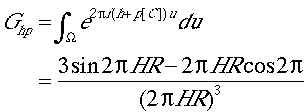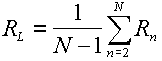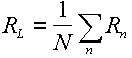![]()
(1)
where
![]()
(2)
and
![]()
![]()
(3)
Given two homologous molecules, A and B, the rotation ([r ]) that brings them into the same orientation will also bring the inter-atomic vectors of each molecule into overlap. Crystallographically, the set of inter-atomic vectors is represented by the Patterson function. Hence, an ordinary rotation function (M. G. Rossmann & D. M. Blow. Acta Cryst. 15, 24--31, (1962)) can be defined as the overlap of one Patterson function and the rotated version of another Patterson function,
|
|
|
(1) |
|
where |
|
|
|
|
|
(2) |
|
and |
|
|
|
|
|
(3) |
[a ] is the deorthogonalization matrix which converts Ångstrom coordinates relative to a Cartesian system to fractional coordinates relative to the crystal unit cell. [b ], the inverse of [a ], is the orthogonalization matrix. The integration volume (W ) is usually a sphere centered at the Patterson origin. The radius (R) of this sphere can be chosen to exclude most of the vectors between atoms in different molecules in the crystal, which are generally longer than the inter-atomic vectors within one molecule.
The rotation [r ] is usually represented by a set of polar (f , y , k ) or Eulerian (q 1, q 2, q 3) angles. In the case of polar angles, f and y define the orientation of a rotation axis relative to a Cartesian coordinate system and k defines the angle of rotation around this axis. In the case of Eulerian angles, the three angles define a sequential set of rotations around the X, Y, Z axes of a Cartesian coordinate system. For both polar and Eulerian angles, the actual meaning of each angle depends on the convention that is used.
The function RF is generally expected to reach its maximum when the rotation [r] corresponds to the rotation that brings the two molecules to the same orientation. In practice, a grid search is carried out covering the entire unique region of the rotational space and Eq. (1) is evaluated at each grid point. The grid point that gives the highest value for Eq. (1) is then possibly the correct rotation. The rotation function values at other grid points that are evaluated in such a search give an estimate for the background noise in the calculation.
Rotation functions can be classified by the way Eq. (1) is evaluated (for example, the slow and the fast rotation functions) or by whether the two molecules reside in the same crystal (self and cross rotation functions).
Eq. (1) can be evaluated in many ways in practice. If both Patterson functions are expressed as their individual Fourier transforms,
|
|
|
|
|
|
it can be shown (M. G. Rossmann & D. M. Blow. Acta Cryst. 15, 24-31, (1962)) that
|
|
|
(4) |
||||||
|
where |
|
|
||||||
|
|
(5)
(6) |
|||||||
is the G function, which represents the Fourier transform of a sphere of radius R. H is the length of the reciprocal space vector h + p [C]. The function assumes the maximum value of 1 when h+p[C]=0 and it quickly approaches and then oscillates around zero as h +p [C] deviates from zero.
Eq. (4) is sometimes referred to as the `slow' rotation function, as its evaluation is generally time-consuming. Several techniques have been developed to speed up the calculations of the slow rotation function. The rotation function value is proportional to the intensity of the reflections (Eq. (4)). It can then be expected that rotation function values are dominated by strong reflections. Therefore, only the strong reflections of crystal B (p) are used in the calculation. These reflections, also known as the large terms (P. Tollin & M. G. Rossmann. Acta Cryst. 21, 872-876, (1966)), are selected based on the criterion
|
|
|
|
where C is the cut-off value and the average intensity < Ip > is generally calculated in shells of equal reciprocal volume. A cut-off value (C) of 1.5 usually selects about 20% of the reflections as large terms.
Given the large terms of crystal B, the summation over the reflections of crystal A (h) can be limited to those which make h + p [C] small, as otherwise the value of the G function will be negligible. For each large term (p) in crystal B, only reflections in crystal A that are within an interpolation box centered on the reciprocal lattice point closest to -p[C] are used in the summation. The size of this interpolation box is generally 3 (reciprocal lattice points) • 3 • 3.
The calculation of the G function value based on Eq. (6) at each rotation search grid point is tedious. Tables of the G function values can be set up beforehand, either in reference to the value of H or in reference to the h, k, l index in the interpolation box, to speed up the calculation.
The values of the rotation function vary relatively slowly as a function of the rotation angles. If a reasonably fine search grid is used, it will be unlikely for the neighbors of a grid point to have high rotation function values if the grid point itself has a low rotation function value. Therefore, the evaluation of rotation functions using Eq. (4) can be accelerated by ignoring such grid points in the calculations (L. Tong. {\it J. Appl. Cryst.} {\bf 26}, 748--751, (1993)).
Even with the improvements described, the calculation of the slow rotation function is still rather time-consuming. The fast rotation function (R. A. Crowther. The Molecular Replacement Method, pp 173-178, Ed. M. G. Rossmann. Gordon & Breach, New York) uses a different approach to evaluate Eq. (1). The Patterson function is expanded in terms of the spherical harmonics,
|
|
|
and
|
|
|
(7) |
|
|
|
(8) |
where jl is the normalized spherical Bessel function of order l and kln is chosen such that jl(kln R) = 0. Ylm is the normalized spherical harmonics function. The expansion coefficient almn can be calculated from
|
|
|
(9) |
where Tlmn is the Fourier transform of Slmn.
With such an expansion for both Patterson functions and utilizing the special properties of the spherical harmonics, Eq. (1) can be simplified to
|
|
(10) |
where the rotation is represented by a set of Eulerian angles (q 1, q 2,q 3), and the function fmm' is only dependent on q 2. Eq. (10) is a two-dimensional Fourier transform over q 1 and q 3. Therefore, for any given q 2, the equation can be evaluated by the fast Fourier transform (FFT) technique, thus greatly accelerating the rotation function calculation.
The calculation of the expansion coefficients almn, however, does still require some amount of computing time. The large term approach as developed for the slow rotation function can also be employed here. Test calculations have shown that a much smaller cut-off (C) value should be used (roughly 0.1 to 0.5) to obtain better rotation function results.
When the two molecules A and B exist in the same crystal, a self rotation function can be used to determine the orientation and the angle of rotation of the local symmetry axis relating the two molecules. Polar angles are usually preferred for representing the rotation [r ] in ordinary self rotation functions. For example, if the molecules are related by a two-fold axis, k can be fixed at 180ƒ and the rotation search can be limited to the f and y angles. The search results can then be presented as a stereographic projection.
If the two molecules are related by an improper rotation (i.e., an angle of rotation not divisible to 360), a full three-dimensional rotation search may be necessary. It is usually more advantageous to represent the rotation [r ] as a set of Eulerian angles in such a case. The unique region of rotational space for different non-cubic space group symmetries is given by S. N. Rao, J.-H. Jih & J. A. Hartsuck. Acta Cryst A36, 878-884, (1980). Once the rotation [r ] is known, a set of polar angles can be extracted from it.
The ordinary rotation function will always have a maximum value at the origin (k = 0 for polar angles, q 2 = 0, q 1 + q 3 = 0 for Eulerian angles) corresponding to the overlap of the Patterson function onto itself. This value can be set to be a specific constant (for example, 1000) and a scale factor is then applied to all the rotation function values to make the maximum 1000. This will bring the self rotation function to an `absolute' scale. The rotations corresponding to the crystallographic symmetry elements should all produce rotation function values of 1000.
If the two molecules are in two seperate crystals, the ordinary cross rotation function can be used to determine the relative orientation of the two molecules. The two crystals may have been prepared experimentally, or an artifical crystal can be prepared by placing a model, usually the structure of a homologous protein, into an arbitrary unit cell. In the latter case, the dimensions of the unit cell are usually chosen large enough so that the inter-molecular vectors are longer than the intra-molecular ones to avoid their interference in the rotation function calculations. Cell dimensions that are twice the dimensions of the model should generally suffice for this purpose.
Eulerian angles are generally preferred for the cross rotation function. The unique region of rotational space for non-cubic space group symmetries of the A and B crystals is given by S. N. Rao, J.-H. Jih & J. A. Hartsuck.
Sometimes it is known that a direction in crystal A is aligned with a direction in crystal B (for example, the alignment of the local two-fold axis of a dimer in two different crystal forms). In such a case, a pre-rotation can be used first to align the two directions. Subsequent rotation searches is then carried out in polar angles, varying only the angle k .
Many macromolecular crystals contain assemblies of the macromolecules obeying a simple (local) point group symmetry. The ordinary self rotation function can be used to determine the orientation of each local symmetry axis individually. However, if the symmetry of the point group is known, orientations of all the symmetry elements of the point group can be determined at the same time, using the so-called locked self rotation function (L. Tong & M. G. Rossmann. Acta Cryst. A46, 783-792, (1990)). It assumes a point group symmetry for the macromolecular assembly and maintains that symmetry throughout its searches (i.e., the local symmetry is locked to the one specified at the outset).
A standard orientation is defined for the local symmetry point group. For example, a 222 point group symmetry can be defined with its three two-fold axes along the X, Y and Z axes of a Cartesian coordinate system. If [In] (n=1, ·, N) represents the set of local symmetry rotation matrices in the standard orientation, the rotation matrices after a rotation [E] has been applied is given by
|
|
|
(11) |
An ordinary self rotation function value (Rn) can be calculated corresponding to each of the rotation matrices. The locked rotation function value (RL) is defined as the average of the individual values,
|
|
|
(12) |
The locked self rotation function calculations are generally carried out in terms of Eulerian angles. In special cases where a local symmetry axis is known to be aligned with a direction in the crystal, polar angles can be used.
The locked rotation function simplifies the self rotation searches for large macromolecular assemblies (for example, viruses). Moreover, it also leads to a reduction, by a factor of about ![]() , in the background noise level of the rotation function as compared to the ordinary self rotation function.
, in the background noise level of the rotation function as compared to the ordinary self rotation function.
The slow rotation function can be employed to calculate the locked self rotation function. Alternatively, the fast rotation function can be used to calculate an ordinary rotation function covering the entire Eulerian space. The locked rotation function can then be calculated by interpolating among the computed ordinary rotation function values.
The locked self rotation function can also be applied to cases where the macromolecular assembly does not obey a simple point group. In such cases, however, it is usually difficult to define a standard orientation.
The presence of local symmetry can also be utilized in the cross rotation searches if only the monomer is used as the model. For every rotation [E] that brings the search model into the same orientation as that of one of the molecules in the assembly, there should be a set of other rotations that will bring the search model into overlap with other molecules in the assembly. Assuming that [Jn] (n=1, …, N) are the local symmetry rotation matrices in crystal B, which can be determined from either ordinary or locked self rotation function calculations, the set of rotations is given by
|
|
|
(13) |
Therefore, a locked cross rotation function can be defined as the average of the individual ordinary cross rotation function values evaluated with the rotation matrices [Cn].
|
|
|
(14) |
Alternatively, assume that [In] (n=1, …, N) are the local symmetry rotation matrices in a standard orientation, [F] is the rotation that brings the standard orientation to that of the assembly in the crystal, and [E] is the rotation that brings the monomer to the same orientation as one of the monomers in the standard orientation,
|
|
|
(15) |
With this definition, it is easier to define the unique region of rotational space for a locked cross rotation function.
Rotation function calculations usually proceed in two steps. In the first step, a search is carried out covering the unique region of the rotational space to find the sets of angles that maximize the rotation function. Self rotation function calculations are usually limited to sections of constant k and the slow rotation function can be used, with grid intervals along f and y of 2 to 3ƒ . For ordinary cross rotation function calculations, the fast rotation function should be used in this initial search. If the results from the fast rotation function is unsatisfactory, the slow rotation function should be tried (but it will take some more CPU time), as it uses a completely different approach. A relatively coarse search grid (generally 3 or 4ƒ intervals) is needed to reduce the total number of grid points in the search.
Once an estimate for the set of angles is obtained, the rotation search should be repeated using finer grid intervals (0.5 to 1ƒ ). The slow rotation function is used for this purpose. The search should be limited to a small region (3 to 5ƒ ) around the values for the rotation angles as determined by the initial search. This will give the optimized values for the rotation angles.
Alternatively, a Patterson-correlation refinement can be used to optimize the angular parameters.
If the local symmetry is a two-fold, the translation element along this axis d// can be determined (M. G. Rossmann, D. M. Blow, M. M. Harding, E. Coller. Acta Cryst. 17, 338-342, (1964)),
|
|
|
(16) |
This calculation does not need the phase information of the reflections.
Once phase information is available, the location of the local symmetry axis in the unit cell can be determined based on the maximal electron-density overlap of the subunits related by the local symmetry (D. M. Blow, M. G. Rossmann, B. A. Jeffrey. J. Mol. Biol. 8, 65-78, (1964); L. Tong. J. Appl. Cryst. 26, 748-751, (1993)).
If sA and sB are the centers of the two molecules,
|
|
|
(17) |
The electron density overlap of the two molecules,
|
|
|
|
where the integration volume is a sphere of radius R centered at sA, can be written as
|
|
(18) |
Given the rotational relationship between molecules A and B and an estimate for the center of molecule A, the center of molecule B can be determined by a Fourier transform (Eq. 18). In this application of Eq. (18), the two molecules can reside either in the same crystal or in two different crystals.
Once initial estimates for [C], sA, sB are available, Eq. (18) can be used to optimize these parameters. A fine search can be carried out around the current values for [C] and sA (or sB, only one of which needs to be varied in the search) to obtain values that will maximize the correlation.
If molecules A and B reside in the same crystal, the position of this local symmetry axis can be determined. Assuming s lies on this axis,
|
|
|
(19) |
where d// is the translation element along this local symmetry axis (see Eq. 16). Eq. 18 then becomes
|
|
|
(20) |
Therefore, the center of the local symmetry axis can be determined by a Fourier transform. This function will produce a long, sausage-shaped density corresponding to the position of the local symmetry axis. To obtain more accurate parameters for the position of the local symmetry element, Eq. (18) should be used. After the maximization of the overlap, the rotational parameters of the local symmetry axis is given by [C] The position of the local symmetry axis is given by (sA + sB) / 2, and d// is given by the projection of sB - sA along the direction of the local symmetry axis.